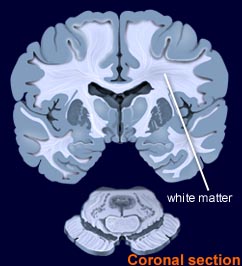
حثل المادة البيضاء
ما هو حثل المادة البيضاء؟
حثل المادة البيضاء هو خلل في الطبقة البيضاء في الدماغ( الدماغ ينقسم بشكل مبسط الى قشرة تسمى المادة الرمادية و اللب و يسمى بالمادة البيضاء) . و هو ليس مرض واحد بل مجموعة كبيرة من الامراض تتجاوز 40 نوع من الامراض الوراثية.
وكلمة حثل يقصد بها تأكل او تهتك.
ماهو المايلين؟
و المخ كبقية جزاء الجسم مكون من بلايين الخلايا منتشرة في عدة اجزاء و تختلف وظيفتها حسب مكان دورها. و الخلية في المخ تسمى خلية عصبية و هي تختلف عن الخلايا الاخرى في الجسم حيث لها امتداد طويل من احد اطرافها يسمى المحور العصبي أو الالياف العصبية وهي تشبه السلك او الكيبل الكهربائي لتوصيل الاشارات الكهربائية من جسم الخلية عبر المحور لأداء المهام في الطرف الاخير للمحور. و هذه الألياف العصبية (المحاور العصبية) مغلفه بطبقة دهنية بيضاء تسمى ( المايلين ) و هذا المادة تعمل على عزل الألياف لتسريع نقل و توصيل الاشارات الكهربائية وعدم خروجها من هذه الالياف الى ان تصل هدفها. بالضبط هي شبيهه بالطبقة البلاستيكية التي تغلف الكيبل الكهربائي لتحمي السلك من الالتماس وتسرع بتوصيل الكهرباء.
المادة الرمادية هي قشرة المخ و فيها تتجمع الخلايا العصبية و تمتد الألياف العصبية من هذه الخلايا الى عمق المخ ( لب المخ).و كثرة هذه الألياف العصبية في داخل لب المخ هي التي تعطي المادة البيضاء لونها الابيض. يتم تصنيع المايلين من خلايا مجاورة للأعصاب تسمى الخلايا قليلة الغصون () ويتم تصنيع مادة المايلين في الخلايا العصبية خارج المخ من خلايا اخرى تسمى خلايا شوان. يتم التحكم في انتاج المايلين من داخل الحمض النووي عبر مجموعة كبيرة من الجينات. عند حدوث خلل في هذه الجينات يحدث تهتك وتدمير متفاوت الشدة لهذه المادة و تظهر على شكل خلل في المادة البيضاء يطلق عليها حثل المادة البيضاء.
ما هي أنواع أمراض حثل المادة البيضاء
يوجد عدد كبير من هذه الأمراض و هي في ازدياد و حاليا يتجاوز عددها 40 نوع. هذا جدول مختصر لهذه الأنواع و بعض اعراضها الأساسية. و في هذا الجدول مجموعة من امراض حثل المادة البيضاء الأكثر شيوعا و شهرتاً. والجدول الذي يليه بقية الأمراض الأقل شيوعا و شهره.
مجموعة من امراض حثل المادة البيضاء الأكثر شيوعا وشهره:
| اسم المرض بالإنجليزية | اسم المرض بالعربي | الأعراض الرئيسية | نمط الوراثة | اسم الجين |
| 18q Syndrome with deficiency of myelin basic protein | متلازمة الذراع الطويلة لكروموسوم 18 المصحوب بنقص البروتين الأساسي للمايلين | غالبا عارض لكن ربما يكون متوارث | ||
| Adrenoleukodystrophy X-Linked (ALD) | حثل المادة البيضاء والغدة الكظرية المرتبط بالجنس | وراثة مرتبطة بالجنس | ABCD1 | |
| Aicardi-Goutieres Syndrome | متلازمة ايكاردي جوتيير | عادتاً وراثة متنحية
واحياناً وراثة السائدة |
TREX1 RNASEH2A RNASEH2B RNASEH2C SAMHD1 ADAR |
|
| Alexander Disease | مرض الاكسندر | وراثة السائدة |
GFAP |
|
| Childhood Ataxia with diffuse CNS Hypomyelination (CACH or Vanishing White Matter Disease) | رنح الأطفال مع نقص المادة البيضاء المنتشر (تلاشي المادة البيضاء) | وراثة متنحية | EIF2B1
EIF2B2 EIF2B3 EIF2B4 EIF2B5 |
|
| Canavan Disease | مرض كانافان | وراثة متنحية | ASPA | |
| Free sialic acid storage disorders 3 | امراض تخزين حمض السيالك | وراثة متنحية | SLC17A5 | |
| Fucosidosis | مرض فكوسيدوسس | وراثة متنحية | FUCA1 | |
| Globoid Cell Leukodystrophy (Krabbe Disease) | حثل المادة البيضاء الكروي الخلوي (مرض كرابي) | وراثة متنحية | GALC See footnote 4 |
|
| L-2-hydroxyglutaric aciduria | ارتفاع حمض الهيدروكسي جلوتاريك |
وراثة متنحية |
L2HGDH | |
| Metachromatic Leukodystrophy (MLD) | حثل المادة البيضاء متبدل اللون (MLD) | وراثة متنحية | ARSA | |
| Oculodentodigital Dysplasia with cerebral white matter abnormalities | الخلل النسيجي للعين والأسنان الأصابع المصاحب لخلل في المادة البيضاء | غالباً
وراثة السائدة واحياناً وراثة متنحية |
GJA1
Peroxisomal acyl-CoA-oxidase deficiency: ACOX1 |
|
| Pelizaeus Merzbacher Disease (X-linked spastic paraplegia) | مرض بليزايوس ميرتسباخر المرتبط بالجنس (الشلل التشنجي المرتبط بالجنس) | وراثة مرتبطة بالجنس | PLP1 | |
| Pelizaeus-Merzbacher-like disease 1 (PMLD1) | المرض المشابه لمرض بليزايوس ميرتسباخر | وراثة متنحية | GJC2 | |
Pol III-related leukodystrophies
|
امراض حثل المادة البيضاء التي تتعلق ببول ثلاثة
نقص المادة البيضاء مع قصور الغدد التناسلية و نقص الاسنان ( متلازمة 4 اتش) مرض الترنح و تاخر ظهور الاسنان و نقص لمايلين بالمخ مرض الارتعاش و الترنح و نقص المايلين بالمخ مرض نقص المايلين بالمخ و ضمور المخيخ و ضمور الكربوس كلوزم |
وراثة متنحية | POLR3A POLR3B |
|
| Refsum Disease | مرض ريفسم | |||
| RNAse T2-deficient leukoencephalopathy | وراثة متنحية | RNASET2 | ||
| Single-enzyme deficiencies of peroxisomal fatty acid beta oxidation | وراثة متنحية | Dibifunctional protein deficiency: HSD17B4 | ||
| Sjogren-Larssen Syndrome | متلازمة شيغرن لارسن | وراثة متنحية | ALDH3A2 | |
| Zellweger Spectrum: Zellweger Syndrome | متلازمة زيلويجر وطيف متلازمة زيلويجر | وراثة متنحية | PEX genes
|
مجموعة من امراض حثل المادة البيضاء الأقل شيوعا وشهره:
| اسم المرض بالإنجليزية | اسم المرض بالعربي | الأعراض الرئيسية | نمط الوراثة | اسم الجين |
| Adult-onset Autosomal Dominant Leukodystrophy (ADLD) | حثل المادة البيضاء المرتبط بالوراثة السائدة للبالغين | وراثة السائدة | LMNB1 | |
| Adult polyglucosan body disease (APBD) | الوراثة المتنحية | GBE1 | ||
| Autosomal Dominant Diffuse Leukoencephalopathy with neuroaxonal spheroids (HDLS) | اعتلال المادة البيضاء الدماغي المنتشر مع تكوير المحاور العصبية المرتبط بالوراثة السائدة | وراثة السائدة | CSF1R | |
| Cerebroretinal microangiopathy w/calcifications & cysts (CRMCC). Coats plus | مرض كوتس بلس | وراثة متنحية | CTC1 | |
| Cerebrtendinous Xanthomatosis (CTX) | مرض التورمات الصفراء الدماغية وفي الأوتار | وراثة متنحية | CYP27A1 | |
| Leukoencephalopathy, cystic, without megalencephaly | اعتلال المادة البيضاء الدماغي الكيسي بسبب خلل في جين RNASET2 | وراثة متنحية | RNASET2 | |
| Leukoencephalopathy w/brain stem & spinal cord involvement & lactate elevation (LBSL) | وراثة متنحية | DARS2 | ||
| Leukoencephalopathy w/thalamus and brain stem involvement & lactate elevation (LTBL) | وراثة متنحية | EARS2 | ||
| Hypomyelination w/atrophy of the basal ganglia & cerebellum (HABC) | نقص المادة البيضاء مع ضمور في العقد القاعدية والمخيخ | وراثة السائدة | TUBB4A | |
| Hypomyelination and congenital cataract (HCC) | نقص المدة البيضاء و الماء الأبيض في العين | وراثة متنحية | FAM126A | |
| Lipomembranous Osteodysplasia with Leukodystrophy (Nasu Disease) | الخلل النسيجي العظمي الغشائي الدهني مع حثل المادة البيضاء (مرض ناسو) | وراثة متنحية | TYROBP
TREM2 |
|
| Megalencephalic Leukodystrophy with subcortical Cysts (MLC) | تضخم الدماغ مع حثل المادة البيضاء و التكيس تحت قشرة المخ (MLC) | وراثة متنحية | MLC1 HEPACAM (MLC2) |
|
| Multiple sulfatase deficiency (MSD) | مرض نقص انزيمات السلفيتيز المتعددة | وراثة متنحية | SUMF1 | |
| Oculodentodigital Dysplasia with cerebral white matter abnormalities | الخلل النسيجي للعين والأسنان الأصابع المصاحب لخلل في المادة البيضاء | غالباً
وراثة السائدة واحياناً وراثة متنحية |
GJA1 | |
| Leukoencephalopathy with dystonia and motor neuropathy | ليكوانسيفالوبثي مع الشد العضلي و ضمور الاعصاب الحركي | وراثة متنحية | SCP2 | |
| SOX10-associated disorders | وراثة السائدة | SOX10 |
الاعراض
المادة البيضاء في الدماغ و التي تحتوي الألياف العصبية هي التي تنقل الإشارات الكهربائية من المخ الى الأطراف لتحريك العضلات. فلذلك اعراض امراض حثل المادة البيضاء تتعلق في البداية بالحركة فيكون هناك تأخر في اكتساب المهارات الحركية بكل أنواعها. كذلك يكون هناك ارتخاء في العضلات يتحسن تدريجيا مع الوقت و لكنه يتحول الى شد و تيبس في العضلات فتصبح صلبه و قاسية يصعب معها تحريك الطفل و فرد اطرافه و قد يبدأ حدوث انحناء و ميلان في العمود الفقري ( الجنف). و عندما تلاحظ الطفل تجد هناك نوع من الشلل في الأطراف و كأنه شلل رباعي او شلل نصفي في الأطراف السفلى. و لان المضغ و البلع و التنفسي يحتاج الى عضلات طبيعية فلذلك تلاحظ ان الكثير من المصابين لديهم صعوبات في التغذية و في التنفس و تكثر لديهم الالتهابات الرئوية مع صعوبات في التنفس نتيجة مشاكل في البلع و دخول الطعام المتكرر للرئة و ضعف في النمو نتيجة لعدم حصولهم على غذاء كافي عن طريق الفم.
كما قد تظهر الاعراض نتيجة لإصابة المخيخ ( و هو الجزء الخلفي من المخ ) و الذي يتحكم بالتوازن . فتظهر اعراضه باهتزاز في العينين و الترنح و ارتجاف و اهتزاز الأطراف مع ارتعاش اليدين و أحيانا حركات لا أراديه مثل الشد و الالتواء العضلي المتكرر.
و تتميز امراض حثل المادة البيضاء بعدم ظهور التشنجات و الصرع في بدايات المرض عكس امراض المادة الرمادية. و لكن قد يظهر الصرع مع تقدم المرض . كما ان القدرات العقلية قد تضعف مع الوقت خاصة في الأنواع التي تصاب البالغين .
هناك اعراض أخرى قد تصاحب امراض حثل المادة البيضاء قد تساعد في التشخيص مثل مشاكل في الاسنان( تأخر بزوغ الاسنان او نقص عدد الاسنان) و ظهور الماء الأبيض في العينين او ضمور في الأعضاء التناسلية او انخفاض في هرمون الغدة الكظرية و الذي قد يظهر على شكل انخفاض في ضغط الدم
أمراض مشابهه لحثل المادة البيضاء:
هناك مجموعة من الأمراض التي تسبب خلل او اضطراب في المادة البيضاء ولكنها لا تصنف على انه حثل في المادة البيضاء. وبعض من هذه الأمراض وراثية و أخرى غير وراثة و نستطيع ان نقسم الأمراض الوراثية منها الى ما يلي:
1- أمراض الأوعية الدموية: و هي مجموعة من الأمراض المختلفة تسبب خلل في الشرايين و العروق الدموية في المخ فتؤثر على المادة البيضاء مثل مرض ” اعتلال الشرايين الدماغية مع الجلطات تحت قشرة المخ و اعتلال المادة البيضاء المرتبط بالوراثة السائدة و المتنحية” و كذلك الامراض المتعلقة بجين COL4A1 و جين COL4A2
2- أمراض تؤثر على المادة البيضاء والرمادية مثل مرض جي أم 1 غانغليوزيدوسس ومرض جي أم 2 غانغليوزيدوسس. و مرض سيرود ليبوفيونوسز العصبي
3- أمراض ارتفاع الأحماض العضوية و الأحماض الأمينية .
4- أمراض الميتوكندريا مثل مرض ميلاس والأمراض المتعلقة بجين بولق
5- أمراض حثل العضلات الخلقي
6- اضطرابات الجليكوزيليشن الخلقية
اما الأمراض التي تصيب المادة البيضاء و لا تصنف على انها أمراض وراثية بحته فهي مثل التصلب اللويحي و اعتلالات المادة البيضاء الناتجة من الالتهابات في المخ و التي تنتج من خلل في جهاز المناعة و هذه الأمراض عادة مكتسبة و ليست بمرض وراثية .
ولكن علينا التنبه ان هناك امراض من حثل المادة البيضاء و هي وراثية بحته و تظهر فقط في البالغين لذلك لا يعني ان الشخص كان سليما في السابق ان المرض ليس من امراض حثل المادة البيضاء او انه غير وراثي. و قد يكون الفيصل في ذلك هو عمل فحوصات وراثية اذا لزم الأمر.
جدول بالأمراض المشابهه لحثل المادة البيضاء
| أسم المرض بالإنجليزي | اسم المرض بالعربي |
| 3-hydroxy-3-methylglutaryl-CoA lyase deficiency | |
| Adenylosuccinase deficiency ACAN-related disorders | الأمراض المتعلقة بالايكان |
| AIMP1-related disorders | الأمراض المتعلقة بالايمب1 |
| Aspartylglucosaminuria | مرض ارتفاع الاسبرتاجليكوزمين في البول |
| Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) | اعتلال الشرايين الدماغية مع الجلطات تحت قشرة المخ و اعتلال المادة البيضاء المرتبط بالوراثة السائدة |
| Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL) | اعتلال الشرايين الدماغية مع الجلطات تحت قشرة المخ و اعتلال المادة البيضاء المرتبط بالوراثة المتنحية |
| Cockayne syndrome and trichothiodystrophy COL4A1-related disorders | متلازمة كوكين |
| Congenital muscular dystrophy | ضمور العضلات الخلقي |
| Defects of N-glycan synthesis including congenital disorders of glycosylation | اضطرابات الجليكوزيليشن الخلقية |
| Dentatorubropallidoluysian atrophy (DRPLA) | |
| Disorders of aminioacidopathy and organic acidemia | أمراض ارتفاع الاحماض العضوية و اضطرابات الأحماض الامينية |
| Disorders of glycoprotein degradation (including alpha-mannosidosis, beta mannosidosis and sialidosis; excluding fucosidosis) | أمراض تحلل الجليكوبروتينات |
| Fabry disease | مرض فابري |
| Familial hemophagocytic lymphohistiocytosis | مرض اكلت الخلايا الوراثي |
| Fatty acid hydroxylase-associated neurodegeneration (FA2H-related disorders) | الأمراض المتعلقة بهيدروكسيليز الأحماض الدهنية |
| Fragile X-associated tremor/ataxia syndrome (FXTAS) | متلازمة كروموسوم أكس لهش |
| Fumarate hydratase deficiency | مرض نقص انزيم فيوميريت هيدريتيز |
| Galactosemia type I | مرض الجلاكتوسيميا النوع الأول |
| Giant axonal neuropathy | |
| Glutaric aciduria type I (GA-I) Glutaric aciduria type II (GA-II; multiple acyl-CoA dehydrogenase deficiency; MADD) | مرض ارتفاع حمض الجليتوريك النوع الأول و الثاني |
| اGlycine encephalopathy | ارتفاع مادة الجليسين |
| GM1 gangliosidosis, infantile onset | مرض جي أم 1 غانغليوزيدوسس |
| GM2 gangliosidosis | مرض جي أم 2 غانغليو |
| infantile onset GPR56-related disorders | |
| Hereditary homocystinurias HSPD1-related disorders | مرض الهوموسيستين يوريا |
| Hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome | متلازمة اتش اتش اتش |
| Hypomelanosis of Ito (HMI; incontinentia pigmenti achromians) | نقص التلون ايتو |
| Incontinentia pigmenti JAM3-related disorders | السلس الصباغي |
| L-2-hydroxyglutaric aciduria | مرض ارتفاع حمض الهيدروكسي جلوترك |
| Lowe syndrome | متلازمة لوي |
| MCT8-specific disorders | مرض خلل جين ام سي تي 8 |
| Menkes disease | مرض مينكي |
| Mitochondrial neurogastrointestinal encephalopathy (MNGIE) | مرض منجي |
| Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke (MELAS) | مرض ميلاس |
| Molybdenum cofactor deficiency and isolated sulfite oxidase deficiency | مرض نقص انزيم معام المليبدم |
| Mucolipidosis IV Mucopolysaccharidosis including MPS type II (Hunter syndrome) | مرض تراكم الدهون المخاطية( النوع الرابع) و السكريات المخاطية ( النوع الثاني) |
| Multiple carboxylase deficiency, including biotinidase deficiency and holocarboxylase synthase deficiency | مرض نقص انزيمات الكربوكسيليز المتعدد |
| Myotonic dystrophy type I | مرض حثل الميوتونيا النوع الأول |
| Neuronal ceroid-lipofuscinoses (NCL) | مرض سيرود ليبوفيونوسز العصبي |
| infantile onset Neuronopathic form of malignant infantile osteopetrosis | مرض تصخر العظام للرضع العصبي |
| Niemann-Pick disease type C | مرض نيمان بيك النوع سي |
| Phenylketonuria (PKU) | مرض الفينايل كيتون يوريا |
| POLG-related disorders | الأمراض الممتعلقة بجين البولج |
| Pyruvate carboxylase (PC) deficiency | مرض نقص انزيم البيروفيت كربوكسيليز |
| Pyruvate dehydrogenase (PDH) deficiency | مرض نقص انزيم البيروفيت ديهيدروجينيز |
| Serine synthesis defects SPG 11 and SPG 15 | مرض تيبس العضلات السفلية النوع 11 و 15 |
| Spondyloenchondrodysplasia | مرض خلل النسيج الفقري الغضروفي |
| Succinic semialdehyde dehydrogenase (SSADH) deficiency (4-hydroxybutyric aciduria) | مرض نقص انزيم السكسينك سميالديهيد |
| Urea cycle disorders: | أمراض حلقة اليوريا |
| Wilson disease | مرض ويلسون |
| Woodhouse-Sakati syndrome (WSS) | متلازمة وودهاوس و سقطي |
التشخيص
ان تشخيص وجود حثل في المادة البيضاء يعتمد على الأشعة المغناطيسية ( اشعة الرنين المغناطيسي) فبدون هذه الاشعة يصعب اكتشاف الخلل في مادة الميلين في تلك الطبقة. و يتم تصنيف امراض حثل المادة البيضاء حسب أجزاء المخ المتأثرة فبعض الامراض تؤثر على الجزء الأمام للمخ اكثر من الجزء الخلف او تؤثر على المخيخ و على قاعدة المخ مع وجود تجاويف او علامات معينة قد تساعد في تحديد المرض المشتبه به .اضافة الى الاعراض الظاهرة على المريض مثل اعراض العيون و الاسنان و الأطراف و غيرها.
لكي يمكن تحديد السبب يتم اجراء فحوصات متعددة مثل تحليل الهرمونات للغدة الدرقية و الكظرية و تحليل بعض المواد الاستقلابية في الدم و البول و قياس بعض الانزيمات في الدم او الجلد و قد يساعد عمل فحص للجينات بشكل مباشرة. و حديثا ظهرت فحوصات تفحص عدة جينات لأمراض حثل المادة البيضاء دفعة واحدة و أخرى بإمكانها فحص جميع الجينات بما يعرف بتسلسل الاكسونات.
حاليا هناك 50% من امراض حثل المادة البيضاء لا يعرف لها جين مسبب و لم يحدد الخلل الوراثي المسبب لها. و هذا يجعل الامر صعبا بخصوص الاستشارات الوراثية و كيفية الوقاية من المرض . و لكن ننصح بالمتابعة مع الأطباء فقط تظهر بعض البحوث نتائج في المستقبل فعلم الوراثة التشخيصي على مستوى فحوصات الجينات في تطور مستمر.
| امراض يصاحبها كبر في حجم الجمجمة | مرض الكسندر و
مرض كنفان مرض ارتفاع حمض الجليتاريك تضخم الدماغ مع حثل المادة البيضاء و التكيس تحت قشرة المخ هناك امراض اخرى وراثية و تسبب كبر في حجم الجمجة لكنها لا تسبب حثل المادة البيضاء و منها: Hexosaminidase A deficiency و Infantile lysosomal storage disorders و Neurocutaneous syndromes مثل PHTS ومرض Hypomelanosis of Ito |
| مشاكل بالجلد | حثل المادة البيضاء والغدة الكظرية المرتبط بالجنس (اسمرار الجلد)
متلازمة شاجرن لارسن (الجلد قشرة السمكة) مرض التورمات الصفراء الدماغية وفي الأوتار |
| مشاكل في العيون: | بقع الكرز الأحمر في الشبكية
مرض جي ام واحد و جي ام ثنين الماء الأبيض بالعين مرض نقص المايلين في المخ و الماء الابيض |
| مشاكل بالأسنان | امراض حثل المادة البيضاء التي تتعلق ببول ثلاثة
مرض الخلل النسيجي للعين والأسنان الأصابع |
العلاج
يعتمد العلاج على نوع المرض و لكن بشكل عام العلاج هو علاج للمشاكل الظاهرة مثل علاج الشد العضلي بادوية تخفف الشد و كذلك علاج الديستونيا و علاج الصرع . كذلك تعويض الجسم بما ينقصه اذا تم اكتشاف بعض النقص مثل نقص الهرمونات . إضافة الى العلاج الطبيعي و حل مشاكل العظام و العمود الفقري التي تطرأ والعلاج التنفسي و التغذية الجيدة و وضع انابيب التغذية اذا لزم الأمر و التأهيل بشكل عام.
نبذه عن أشهر أمراض حثل المادة البيضاء:
حثل المادة البيضاء متبدل اللون
هذا المرض مرض وراثي متنحي ناتج عن نقص احد الانزيمات المهمة للمادة البيضاء في المخ. و يعتبر من الأمراض النادرة لكنه اكثر امراض حثل المادة البيضاء شيوعاً .و يتميز بتراكم تراكم مادة دهنية تعرف باسم سلفاتيد (أ الشحميات السفينغولية) في الدماغ ومناطق أخرى من الجسم (مثل الكلى و الكبد والطحال و المرارة). يؤدي تراكم مادة السلفا هذه الى فقدان الغطاء الواقي الدهني الذي يغلف الألياف العصبية (المايلين) من المخ .وتشمل أعراض حثل المادة البيضاء متبدل اللون تأخر النمو شد في العضلات يزداد مع العمر و التشنجات، و الصرع، وتغيرات سلوكية في الشخصية، ، و فقدان القدرات العقلية تدريجياً، وضعف البصر .
و هناك ثلاثة انواع المرض حسب سن ظهور الاعراض و حسب مستوى النقص في الانزيم .
الأطفال: و تظهر الاعراض منذ الولادة تقريبا
الأحداث : وتظهر الاعراض في سن5 الى 7 من العمر
البالغين: تظهر في بدايات سن المراهقة بعد سن 15 سنة من العمر.
الأعراض:
العلامات والأعراض مبكرة المصاحبة لهذا المرض قد تكون في البداية غامضة وتدريجي في البداية، مما يجعل التشخيص صعب . و اول الاعراض التي تلاحظ هي تغير في التفكير و الذاكرة، وتغير في وضعية الجسم الحركية .. أحيانا تكون أول الاعراض تغير في النظر و صعوبة في الرؤية أو خدر و تنميل في بعض اجزاء الجسم.
في النوع الذي يصيب الرضع تظهر الأعراض في اواخر السنة الأولى الى سن الثانية او الثالثة من العمر، قد تكون الأعراض الأولية التهيج و القلق ، ارتخاء العضلات، واضطرابات في المشية. بينما تظهر الأعراض في النوع الذي يصيب الأحداث بين سن 4 و 10 سنوات من العمر .و . النوع الذي يصيب البالغين يبدأ بعد 15 سنة من العمر و في معظم الحالات خلال العقد الثالث أو الرابع من العمر. الأعراض في جميع أشكال المرض متشابهة. و لكن قد تكون الصعوبات البصرية أكثر وضوحا في الأطفال الرضع، في حين ان تدهور القدرات العقلية و الذهان والخرف قد يكون أكثر وضوحا عند البالغين المصابين بهذا المرض. لكن من المهم معرفة ان الأعراض قد تتفاوت بشكل كبير بين الأفراد المصابين.
ويمكن أن تشمل الأعراض كما ذكرنا الى مشاكل في البصر مما يؤدي إلى العمى، تغيرات في الشخصية، والاضطرابات الحركية مثل عدم التوازن في المشي و الترنح، وضعف و ارتخاء العضلات مع حدوث موجات من تصلب العضلات و شدتها و عدم القدرة على تنسيق حركة (الاهتزاز)، و تقلصات العضلات الى الخلف خاصة ل الرقبة والعمود الفقري والذراعين والساقين. ويمكن أن تشمل الأعراض الأخرى انتفاخ في البطن، صعوبة في الكلام (تأخر النطق)، وفقدان المهارات الذهنية التي اكتسبها الطفل سابقا، و التشنجات.
و كون مادة المايلين التي تغطي الألياف العصبية في المخ و ايضا تغطي الألياف العصبية في الأطراف ففد تظهر اعراض غامضة في الأطراف نتيجة تأثر الاعصاب الحركية الطرفية و الحسية مثل ضعف العضلات و التنميل و الألم و الاحمرار خاصة في الساقين و اليدين.
الأسباب
مرض حثل المادة البيضاء متبدل اللون مرض من الامراض الوراثية التي تنتقل بما يعرف بالوراثة المتنحية. حيث يكون كلا الابوين ناقلين للمرض و لكنهما سليمين و ليس لديهم اي من اعراض المرض. و لكن قد يتكرر المرض في كل مره تحمل فيها الأم بنسبة 25% واحتمال عدم تكرارها 75% و لكن بعض الاطفال السليمين قد يكونوا ايضا ناقلين للمرض مثل ابويهم و بعضهم سليم بالكامل. راجع صفحة الورثة المتنحية في موقع الوراثة الطبية.
الجين المعطوب هو الذي ادى الى نقص في الانزيم الذي يمنع تراكم مادة السلفا في الميلين في الاعصاب نوع A . يقع هذا على الذراع الطويلة من كروموسوم 22 . و حسب شدة انخفاض مستويات هذا الإنزيم تظهر الاعراض في الرضع او في الاحداث او البالغين حيث يكون النقص شديد الا كامل في حال النوع الذي يصيب الرضع بينما يكون النقص متوسط في النوع الذي يصيب الاحداث و يكون خفيف في النوع الذي يصيب البالغين. هناك جين اخر تم اكتشافه يسبب نفس المرض اسمة ( SAP-B ) ويقع على الذراع الطويلة من كروموسوم 10 . و الأعراض السريرية لهذا الجين مماثلة لأعراض الجين الاخر و يشكل في انها السبب اذا كان مستوى الجين الاول سليم و تحليل البول يظهر ارتفاع في مادة السلفاتيد . و هذا الجين ينتج انزيم محفز لتفكيك الدهون و لها علاقة في المايلين في المادة البيضاء في المخ .
نسبة انتشاره
يعتبر هذا المرض من الامراض النادية و يصيب الذكور والإناث بالتساوي .و يصيب كل الشعوب و الاعراق .نسبة انتشار النوع الطفولي حوالي طفل لكل 40 الف طفل بينما النوع الذي يصيب الأحداث طفل مصاب لكل 15 الف طفل .
التشخيص
يتم التشخيص المبدئي عن طريق اشعة الرنين المغناطيسي للمخ حيث يتم توثيق وجود حثل في المادة البيضاء و يتم الشك في وجود هذا المرض عن طريق قياس مادة السلفاتيد في البول يتبعها قياس مستوى الانزيم في الدم او الجلد و يتم بعدها فحص الجين لاكتشاف الجين و الطفرة المسببة للمرض.
العلاج
رغم انه لا يوجد علاج شافي من المرض الى وقتنا الحالي الا ان العلاج يتمثل في علاج الاعراض كعلاج الصرع و تحسين التغذية و العلاج الطبيعي لتقليل تيبس المفاصل . في النوع الذي يصيب الاحداث هناك محاولات بحثية لعلاج المرض عن طريق زراعة نخاع العظم بالخلايا الجذعية .
الوقاية
بالإمكان عمل فحص للجنين خلال الحمل في حال وجود حالات اصابة سابقة. و يمكن قياس مستوى الانزيم في الخلايا المستذرعه من السائل الذي يحيط بالجنين ( ماء الجنين المعروف بالسائل الامينوسي). او فحص الجين المسبب للمرض في حال معرفة الطفرة المسببه للمرض في الاسرة
امراض مشابهة لمرض
مرض الغدة الكظرية و حثل المادة البيضاء:
هو مرض وراثي نادر يتميز بفقدان الغطاء العازل الدهني (المايلين) الذي يغلف الألياف العصبية في يصاحبه ضمور تدريجي للغدة الكظرية (الغدة فوق الكلوية). لهذا المرض عدة اشكال لكن اكثرها شيوعا من النوع الذي يحدث خلال مرحلة الطفولة. و تشمل الأعراض: التغيرات السلوكية مثل ضعف الذاكرة، ، وفقدان السيطرة على المشاعر تدهور التحصيل الدراسي في المدرسة و تدهور القدرات العقلية. ويمكن أن تشمل الأعراض ايضاً ضعاف القدرة على تنسيق الحركة (ترنح و الاهتزاز و ارتخاء وضعف العضلات و قد يكون في احد جانبي الجسم ( كحالات الشلل النصفي )،و صعوبات في الكلام و النطق، وضعف و فقدان السمع، و مصاعب بصرية. يصاحب هذا المرض ارتفاع في نسبة بعض المواد الدهنية في الدم و التي تعرف باسم الأحماض الدهنية طويلة السلسلة (VLCFA) .وهو مرض يصيب الذكور و ينتقل بالوراثة المرتبطة بالجنس في اشهر انواعه .(للمزيد من المعلومات حول هذا المرض اضغط هنا)
المرض الإسكندر
هو مرض ناتج عن خلل وراثي و في العادة غير متوارث . وو نادر جداً و هو من اندر امراض حثل المادة البيضاء و يتميز بفقدان الغطاء العازل الدهني (المايلين) الذي يغلف الألياف العصبية. أعراض هذا الاضطراب عادة ما تبدأ في مرحلة الطفولة ويمكن أن تشمل شد في العضلات، وتأخر النمو و اكتساب المهارات، تشنجات و تأخر عقلي. و عندما يبدأ ظهور المرض خلال مرحلة الطفولة، فان الأعراض تشمل صعوبة في البلع وآلام في المفاصل، والتقيؤ المتكرر، وصعوبة في التنفس وعدم القدرة على السعال، و الشد العضلي. كما يتميز هذا المرض عن بقية امراض حثل المادة البيضاء بكبر حجم الجمجمة .كما انه يصيب الذكور اكثر من الاناث .و في العادة يكون المصاب طفل واحد في الاسرة لان سببه طفرة جديدة. و لكن هناك بعض الحالات الوراثية خاصة التي تظهر فيها الاصابة في البالغين .و تم حديثا التعرف على الجين المسبب لهذا المرض و بالامكان عمل فحص جيني للمصاب و لوالديه.(للمزيد من المعلومات حول هذا المرض اضغط هنا).
مرض كانافان
هو مرض وراثي نادر يتميز بفقدان الغطاء العازل الدهني (المايلين) الذي يغلف الألياف العصبية في المخ و الحبل الشوكي. و هو مثل مرض الاسنكدر يتميز بكبر حجم الجمجمة. و تشمل أعراضه ارتخاء وضعف في العضلات ، وفقدان الطفل للمهارات التي سبق أن اكتسبها كانت عقلية او حركية مع ضعف التحكم الرقبة و ضعف البصر الذي قد يؤدي الى العمى. كما يمكن أن تشمل الأعراض التقلصات اللاإرادية في العضلات في الذراعين والساقين. قد تبدأ الأعراض في مرحلة الطفولة المبكرة، وعادة ما تتطور بسرعة، مما يؤدي إلى مضاعفات تهدد الحياة. .(للمزيد من المعلومات حول هذا المرض اضغط هنا).
مرض كرابيه (حثل المادة البيضاء كرابيه)
هو مرض وراثي نادر جداً يتميز بفقدان الغطاء العازل الدهني (المايلين) الذي يغلف الألياف العصبية بسبب تراكم غير طبيعي لمادة دهنية في الدماغ تسمى السيراميد جلكتوسيد. و تشمل الأعراض التهيج والتقيؤ، ونوبات من فقدان الوعي الجزئي و التشنجات.كما قد يكون يصاحبها تقلصات وشد في الساقين، وصعوبة في البلع، و تدهور القدرات العقلي. .(للمزيد من المعلومات حول هذا المرض اضغط هنا).
مرض تاي ساك
هو مرض نادر وراثي تدريجيا يسبب ضعف في القدرات العصبية للدماغ . و هو خلل في تعامل الجسم مع الدهون وذلك بسبب نقص او فقدان انزيم مهم يسمى (هيكسوزامينيداز الوحده أ ). مما يؤدي الى تراكم هذه الدهون في الدماغ. و تشمل الأعراض نووبات الفزع مثل ما يحدث عند ترويع الشخص بشكل مفاجئ و تتكرر هذه الحركات بشكل متكرر. كما يصاحبه تشنجات و شد في العضلات و حركات لاإرادية للأطراف بشكل متكرر . و قد تظهر اعراض اضافية ما بين 6 و 10 شهرا من العمر، مثل صعوبات التغذية، وارتخاء و ضعف العضلات (نقص التوتر)، والأرق و القلق ، وحركات العين الغير عادية، وقد يشاهد طبيب العيون بجهاز فحص الشبكية بقع دائرية حمراء تسمى ( بقع الكرز الحمراء). بعد سن 12 شهرا قد يفقد الطفل المهارات التي اكتسبها سابقا. يشتهر هذا المرض في شعوب اوروبا الشرقية و في الاصول اليهودية و لكن هناك اصابات من شعوب اخرى.
مرض ساندهوف
و مرض نادر وراثي تدريجيا يسبب ضعف في القدرات العصبية للدماغ . وهو مشابه لمرض تاي ساك لكنه الاعراض فيه اشد و يصيب كل الشعوب. و هو ناتج عن خلل في تعامل الجسم مع الدهون وذلك بسبب نقص او فقدان انزيم مهم (هيكسوزامينيداز الوحده ب ). الأعراض الأولى للمرض تبدأ بين 3 إلى 6 أشهر من العمر و تشمل تأخر في النمو معض ضعف و ارتخاء في العضلات ة وحركات فزع متكررة مع مشاكل في التغذية، وقد يشاهد طبيب العيون بجهاز فحص الشبكية بقع دائرية حمراء تسمى ( بقع الكرز الحمراء). وتشمل الأعراض الأخرى التدهور العقلي التدريجي العقلية، التشنج و الصرع، و الحركات اللاإرادية للأطراف و مشاكل في دقات القلب، و الضعف البصري و قد يتضخم الطحال بشكل غير طبيعي.
مرض بليزايوس-ميرتسباخر
هو مرض وراثي نادر جداً يتميز بفقدان الغطاء العازل الدهني (المايلين) الذي يغلف الألياف العصبية. قد تبدأ الأعراض في مرحلة الرضع (في النوع التقليدي المشهور للمرض) أو في مرحلة البلوغ. عند الرضع، تشمل أعراض تأخر النمو و فشل التحكم في حركة الرأس والعينين، ، ارتعاش الاطراف و حركات لا ارادية مع تقطيب في حركة عضلات الوجه، ، وضعف، عام مع عدم القدرة على الثبات عند الوقوف يتبعه تيبس و تقلصات في المفاصل . .(للمزيد من المعلومات حول هذا المرض اضغط هنا).
مرض الغدة الكظرية و حثل المادة البيضاء:
هو مرض وراثي نادر يتميز بفقدان الغطاء العازل الدهني (المايلين) الذي يغلف الألياف العصبية في يصاحبه ضمور تدريجي للغدة الكظرية (الغدة فوق الكلوية).
لهذا المرض عدة اشكال لكن اكثرها شيوعا من النوع الذي يحدث خلال مرحلة الطفولة. و تشمل الأعراض: التغيرات السلوكية مثل ضعف الذاكرة، ، وفقدان السيطرة على المشاعر تدهور التحصيل الدراسي في المدرسة و تدهور القدرات العقلية. ويمكن أن تشمل الأعراض ايضاً ضعاف القدرة على تنسيق الحركة (ترنح و الاهتزاز و ارتخاء وضعف العضلات و قد يكون في احد جانبي الجسم ( كحالات الشلل النصفي )،و صعوبات في الكلام و النطق، وضعف و فقدان السمع، و مصاعب بصرية. يصاحب هذا المرض ارتفاع في نسبة بعض المواد الدهنية في الدم و الجهاز العصبي و التي تعرف باسم الأحماض الدهنية طويلة السلسلة (VLCFA) .وهو مرض يصيب الذكور و ينتقل بالوراثة المرتبطة بالجنس في اشهر انواعه .فلذلك هو ناتج عن عطب ( طفرة) في احد الجينات الموجودة على كروموسوم أكس .فمن المعروف ان كل الاناث لديهن نسختين من كروموسوم أكس بينما الذكور لديهم فقط نسخة من أكس و اخرى من كروموسوم واي. فاذا حدث عطب او خلل في احد نسخ هذا الجين التي عند المرأة فانه في العادة لا تظهر عليها اعراض للمرض لان هناك نسخة اخرى تعمل. بينما الذكور اذا حدث عطب في النسخة الوحيدة التي لديهم فان المرض يظهر عليهم.
هناك عدة اشكال للمرض و تتفاوت في شدتها و اعراضها و وقت ظهورها. اشهرها هو الذي يحدث للأطفال و المعروف بحثل المادة البيضاء و يمثل حوالي 45% من الحالات و يتميز بفقدان و تأكل في مادة المايلين و يزيد مع الوقت و لذلك تزداد الاعراض المتعلقة بالجهاز العصبي تدريجيا و بلا هوادة و قد تصل لحالة فقدان الوعي و قد تؤثر على الحياة في غضون خمس سنوات كمعدل.
الأعراض
تتفاوت الأعراض حسب عمر الشخص و جنسه و انسجة الجسم التي تأثرت بالمرض.. و اكثر الانسجة التي تتأثر في الجسم هي مادة الميلين التي تغطي الالياف العصبية و خلايا الدم و الغدة الكظرية( فوق الكلوية).قد لا تتأثر هذه الاعضاء و الانسجة في نفس الوقت في جميع المرضى . قام الاطباء بتقسيم هذا المرض حسب الاعضاء و الانسجة التي تتأثر و تم تصنيف اربع انواع رئيسية منها لكن علينا ان نعرف ان المريض قد تظهر عليه بعض منها حتى و ان تم تصنيف مرضه بأحدها بشكل خاص.
النوع الأول: بدون اعراض
النوع الثاني : اعراض ضمور الغدة الكظرية و ضعف الأعصاب
النوع الثالث : ضمور الغدة الكظرية فقط
النوع الرابع : حثل المادة البيضاء فقط
النوع الأول و برغم ان الشخص مخبريا لديها عطب في الجين لكن لا تظهر عليه الاعراض .و هذا يكثر في الاناث مقارنة بالذكور . و معظمهم لا تظهر عليه الاعراض في اول ثلاث سنوات من العمر. و لكن من الملاحظ انه مع الوقت قد تظهر بعض الاعراض و هناك من يستمر بدون اعراض بقية عمره.
النوع الثاني تكون اعراضه خليط من اعراض ضمور و ضعف الغدة الكظرية و من الاعراض العصبية مثل ضعف العضلات ثم تصلب و شد في عضلات الساقين و صعوبة في المشي و عدم التوازن و الترنح كما قد تظهر اعراض تنميل في الاطراف و ضعف خفيف في الاطراف العليا و قد يكون مصحوب بمشاكل في التبول و ضعف جنسي.
و النوع الثالث تكون الاعراض هي اعراض ضمور الغدة الكظرية و هي اعراض قد لا تكون واضحة مثل الضعف العام و التعب و اللام البطن و لكن من اهم الاعراض هو اسمرار البشرة و انخفاض ضغط الدم و السكر من غير سبب واضح.
والنوع الرابع قد تظهر اعراضه في الطفل الذكر على شكل مشاكل واخفاقات دراسية مع عدم القدرة على التركيز و مسك القلم بشكل جيد مما قد يكون هناك صعوبة في النطق و الكلام مع ان الطفل يفهم ما يقال له. كما قد يجد الطفل صعوبة في القراءة و فهم ما يقرأه مع صعوبات ف التوازن و مشاكل في البصر و أحيانا ازدواج في البصر و ظهور سلوكيات غير معتادة مثل العصبية و العدوانية. في المقابل قد يكون الصرع هو اول اعراض المرض في بعض الأولاد. كما قد تتفاوت الاعراض حتى في الاسرة الواحدة و حتى في التوائم المتطابقة. من الاعراض التي ينبغي التنبه لها ما يلي:
• المشاكل السلوكية
• فرط النشاط
• اضطراب نقص الانتباه
• ألم في العين / الصداع النصفي الذي يظهر في الطفولة
• الالتهابات الفيروسية المتكررة
• الخمول، التعب بسهولة، عدم التوازن
• نقص السكر في الدم
• اسمرار الجلد
• قصور الغدة الكظرية
التشخيص
يعتمد التشخص على التنبه للأعراض ونتائج الفحوصات الأولية مثل قياس مستوى الدهون طويلة السلسلة. فهي معظم الوقت مرتفعة في حالة الذكور المصابين بينما الاناث قد تكون في المستوى الطبيعي. كم يتم تأكيد التشخيص بفحص الجين المسبب للمرض (ABCD1). ويمكن فحص أي فرد من افراد الاسرة يشتبه بإصابته عن طريق فحص الجين والتأكد ما اذا كانت الطفرة المسببة للمرض موجوده ام لا.
الأسباب
ينتج هذا المرض عن نقص اما كامل او جزئي في انزيم مهم ينتجه أحد الجينات الموجودة على كروموسوم أكس. هذ الجين اسمه (ABCD1). و هو ينتقل من الام الى اطفالها الذكور خاصة لان الام تعطي كروموسوم أكس و الاب يعطيهم كروموسوم واي. ولكن أيضا بعض البنات قد تصاب اذا حصلت على الجين المعطوب من أمها. راجع الوراثة المرتبطة بالجنس في موقع الوراثة الطبية لمزيد من التفصيل.
العلاج
ليس هناك علاج شافي دوائي. لكن قد تكون عملية زراعة نخاع العظم هي الحل المتوفر حاليا.كما وجد ان زيت لورنزو (Lorenzo’s Oil) يفيد بعض الحالات فيقلل و يأخر من ظهور الاعراض. كما ان استعمال حمية غذائية خاصة تخفض مستوى الدهون ذات السلسلة الطويلة و قد تفيد بعض الحالات.
تأليف
الدكتور عبدالرحمن فايز السويد
استشاري الأمراض الوراثية الإكلينيكية و طب الأطفال
السلام عليكم
عندي طفلين مصابين بمرض نقص المادة البيضاء ويتناولون علاج تقريتول وباكلوفين ورفترين اريد اي معلومة تفيدني
السلام عليكم انا عندي بنت ظهر في التحاليل نرض المادة البيضه اذا لقات علاج وفاد عيالك فا جزاك الله دلنا عليه وتكسب الاجر هذا رقم تليفوني 00971508244477
السلام عليكم انا عندي بنت ظهر في التحاليل نرض المادة البيضه اذا لقات علاج وفاد عيالك فا جزاك الله دلنا عليه وتكسب الاجر هذا رقم تليفوني 00971508244477
وعليكم السلام
لعل المعلومات بالموقع تفيدك باذن الله
عندى ابنى مصاب بمرض وراثى هو mpstype2 (hunter syndrome) من امراض التمثيل الغذاءى فهل لهو علاج ولمعرفة تاثيره على المخ اعمل رنين مغناطيسى ارجو الرد
Pingback: رسائل 2018 | مجموعة الدعم الأسري لأمراض التمثيل الغذائي
Pingback: رسائل 2016 | مجموعة الدعم الأسري لأمراض التمثيل الغذائي